Dr. Thanet Pitakbut's Biography

Molecular simulation, AI, and Drug Discovery research
Go back to the first page Click
Utilizing AI-based QSAR model and molecular docking for anti-bacterial drug discovery 💻💊
Motivation:
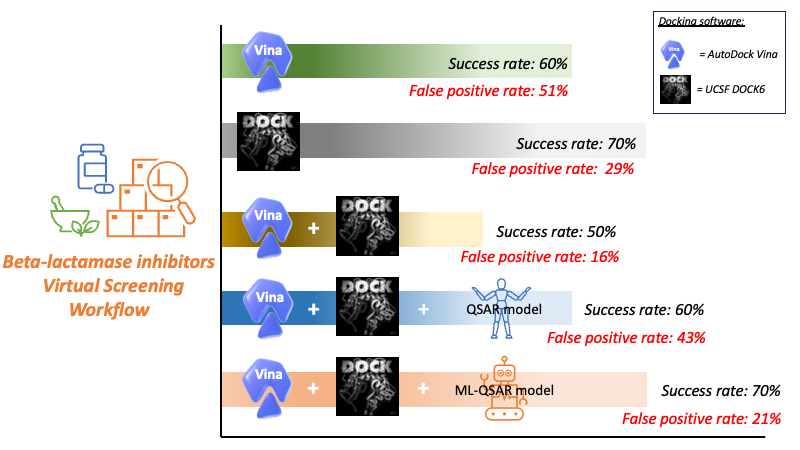
The fast development of antimicrobial-resistant bacteria is one of the factors pressuring pharmaceutical research and development, and it is commonly known that beta-lactamase is one of the common resistance mechanisms in bacteria and significantly contributes to an ongoing global health problem. In this regard, beta-lactamase is selected and used as a drug target. In current digital drug discovery research, molecular docking is one of the most common computational simulation methods used in the field. Recent studies have shown that combining AI with molecular docking simulation can improve drug discovery performance. Therefore, in this project, AI-based QSAR (Quantitative Structure-Activity Relationship) is integrated with molecular docking to enhance anti-lactamase inhibitor search.
Highlight:
- An optimized docking approach can maximize the success in identifying beta-lactamase by up to 70%.
- Standard consensus docking sacrifices part of the accuracy to minimize the false positive by half.
- Integrating the AI-based QSAR model into molecular docking restores the success rate of consensus docking while maintaining a low false positive rate.
Further reading:
Pitakbut, T., Munkert, J., Xi, W., Wei, Y. and Fuhrmann, G., 2024. Utilizing machine learning-based QSAR model to overcome standalone consensus docking limitation in beta-lactamase inhibitors screening: a proof-of-concept study. BMC Chemistry, 18(1), p.249.
Read the article [Click here!]
